 درمانگاه شبانهروزي عمومی و تخصصي فاضل با هدف ارتقاي سلامت مردم منطقه شمال شهر اصفهان در سال 1395 به صورت خیریه فعاليت خود را آغاز نموده است.
درمانگاه شبانهروزي عمومی و تخصصي فاضل با هدف ارتقاي سلامت مردم منطقه شمال شهر اصفهان در سال 1395 به صورت خیریه فعاليت خود را آغاز نموده است.
رتینیت پیگمانتوزا
رتینیت پیگمانتوزا (Retinitis Pigmentosa یا RP) یک گروه از بیماریهای ارثی شبکیه است که منجر به تحلیل تدریجی سلولهای حساس به نور در شبکیه میشود. این بیماری به مرور زمان باعث کاهش دید محیطی و دید در شب میشود و در مراحل پیشرفته میتواند به کوری کامل منجر شود.
علائم رتینیت پیگمانتوزا
به مرور زمان و با پیشرفت بیماری تغییر میکنند. این علائم معمولاً در دوران کودکی یا نوجوانی آغاز میشوند و شامل موارد زیر هستند:
- شبکوری:
- کاهش توانایی دید در نور کم یا تاریکی، که اولین و شایعترین علامت RP است.
- کاهش دید محیطی (تونلبینی):
- از دست دادن دید محیطی یا دید جانبی، که باعث میشود فرد فقط بتواند به طور مستقیم به جلو نگاه کند و دید محیطی محدود شود.
- کاهش تدریجی وضوح دید مرکزی:
- با پیشرفت بیماری، دید مرکزی نیز ممکن است کاهش یابد، که این امر به ویژه در مراحل پیشرفته بیماری دیده میشود.
- اختلال در تشخیص رنگها:
- کاهش توانایی تشخیص رنگها و دیدن رنگها به درستی، که ممکن است در مراحل پیشرفتهتر بیماری رخ دهد.
- دیدن نقاط تیره یا نورهای چشمکزن:
- افراد ممکن است نقاط تیره یا نورهای چشمکزن در میدان دید خود ببینند.
- حساسیت به نور (فوتوفوبیا):
- افزایش حساسیت به نور روشن که ممکن است باعث ناراحتی و مشکل در دیدن در نور زیاد شود.
علائم جزئیتر و موارد دیگر:
- نوسانات دید: گاهی ممکن است دید بیمار به صورت موقت بهبود یابد یا بدتر شود.
- تجمع رنگدانهها در شبکیه: چشمپزشکان ممکن است با معاینه چشم، تجمع غیرطبیعی رنگدانهها در شبکیه را مشاهده کنند، که یکی از نشانههای RP است.
با توجه به این که رتینیت پیگمانتوزا یک بیماری پیشرونده است، علائم ممکن است به مرور زمان بدتر شوند. افرادی که علائم اولیه RP را تجربه میکنند باید به یک چشمپزشک مراجعه کنند تا تشخیص دقیقتری انجام شود و برنامهریزی برای مدیریت بیماری صورت گیرد.
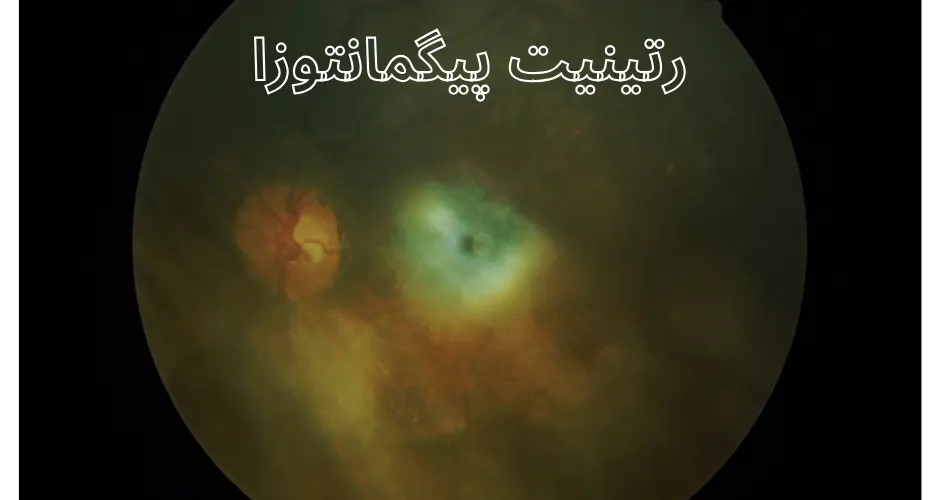
علل رتینیت پیگمانتوزا
رتینیت پیگمانتوزا (Retinitis Pigmentosa یا RP) یک بیماری ژنتیکی است که به دلیل جهشهای مختلف در ژنهای مرتبط با عملکرد و ساختار شبکیه چشم ایجاد میشود. این جهشها باعث تخریب تدریجی سلولهای حساس به نور در شبکیه میشوند. علت اصلی RP معمولاً به یکی از سه الگوی وراثتی زیر مرتبط است:
۱. وراثت اتوزومال غالب
- ویژگیها: فقط یک نسخه از ژن معیوب از یکی از والدین کافی است تا بیماری به فرد منتقل شود.
- احتمال انتقال: ۵۰% احتمال انتقال بیماری به فرزندان وجود دارد.
- شدت بیماری: علائم ممکن است کمتر شدید باشند و پیشرفت بیماری ممکن است کندتر باشد.
۲. وراثت اتوزومال مغلوب
- ویژگیها: دو نسخه از ژن معیوب (یکی از هر والد) برای بروز بیماری لازم است.
- احتمال انتقال: ۲۵% احتمال دارد که فرزند به بیماری مبتلا شود اگر هر دو والد ناقل باشند.
- شدت بیماری: علائم ممکن است شدیدتر باشند و بیماری معمولاً در اوایل دوران کودکی یا نوجوانی ظاهر میشود.
۳. وراثت وابسته به جنس (X-linked)
- ویژگیها: ژن معیوب بر روی کروموزوم X قرار دارد. مردان (با یک کروموزوم X) بیشتر تحت تأثیر قرار میگیرند.
- احتمال انتقال: مردانی که ژن معیوب را به ارث میبرند حتماً بیماری را نشان میدهند. زنان ناقل ممکن است علائم خفیفتری نشان دهند یا حتی بدون علائم باشند.
- شدت بیماری: مردان مبتلا معمولاً علائم شدیدتری دارند و بیماری سریعتر پیشرفت میکند.
ژنهای مرتبط با RP
بیش از ۶۰ ژن مختلف تا کنون شناسایی شدهاند که جهشهای آنها میتواند به RP منجر شود. برخی از ژنهای شایع مرتبط با RP عبارتند از:
- ژنهای رودهپسین (RHO)
- ژنهای پرایموردیال دیسکینزی (PRPH2)
- ژنهای یوستن (USH2A)
- ژنهای آلفا-توبولین (TUB)
سایر عوامل ژنتیکی
- موزاییسم ژنتیکی: جهشهای جدید در سلولهای زایشی والدین که به فرزندان منتقل میشود.
- تغییرات در نواحی تنظیمی ژن: برخی جهشها ممکن است در نواحی غیر کدکننده ژنها رخ دهند که بر بیان ژنها تأثیر میگذارند.
نقش غیر ژنتیکی
در برخی موارد نادر، عوامل محیطی و تغذیهای ممکن است بر پیشرفت و شدت بیماری تأثیر بگذارند، اما علت اصلی همچنان ژنتیکی است.
پیشگیری
پیشگیری از RP به دلیل ماهیت ژنتیکی آن ممکن نیست، اما مشاوره ژنتیکی برای افرادی که سابقه خانوادگی این بیماری را دارند، میتواند به درک بهتر خطر انتقال بیماری به نسلهای بعدی کمک کند.
تشخیص زودهنگام و مدیریت مناسب میتواند به بهبود کیفیت زندگی بیماران مبتلا به RP کمک کند. افراد مشکوک به این بیماری باید به یک چشمپزشک و متخصص ژنتیک مراجعه کنند.
تشخیص
معمولاً شامل ترکیبی از ارزیابیهای بالینی، آزمایشهای تخصصی چشم، و در برخی موارد آزمایشهای ژنتیکی است. مراحل تشخیص به شرح زیر هستند:
۱. تاریخچه پزشکی و خانوادگی
- بررسی علائم: پزشک از بیمار درباره علائم مشاهده شده، مدت زمان بروز آنها و تغییرات اخیر سوال میکند.
- سابقه خانوادگی: پزشک درباره وجود بیماریهای مشابه در خانواده سوال میکند، زیرا RP یک بیماری ژنتیکی است.
۲. معاینات بالینی چشم
- معاینه با اسلیت لامپ: برای بررسی جلوی چشم و تشخیص هرگونه تغییرات.
- معاینه ته چشم (افتالموسکوپی): برای بررسی شبکیه و عصب بینایی. پزشک به دنبال علائم مشخصه RP مانند تجمع رنگدانههای غیرطبیعی، نازک شدن عروق شبکیه و تغییرات در عصب بینایی میگردد.
۳. آزمایشهای تخصصی چشم
- الکترورتینوگرافی (ERG): این آزمایش پاسخهای الکتریکی شبکیه به نور را اندازهگیری میکند. افراد مبتلا به RP معمولاً کاهش قابل توجهی در پاسخهای الکتریکی شبکیه دارند.
- آزمایش میدان دید: این آزمایش دید محیطی بیمار را ارزیابی میکند. افراد مبتلا به RP اغلب کاهش دید محیطی (تونلبینی) دارند.
- تصویربرداری از شبکیه: روشهایی مانند تصویربرداری با استفاده از توموگرافی نوری همدوسی (OCT) و فلوروسین آنژیوگرافی برای بررسی ساختار شبکیه و جریان خون استفاده میشود.
۴. آزمایشهای ژنتیکی
- تحلیل ژنتیکی: برای شناسایی جهشهای ژنتیکی مرتبط با RP انجام میشود. این آزمایش میتواند به تایید تشخیص و شناسایی نوع خاص RP کمک کند.
- مشاوره ژنتیکی: بیماران و خانوادههای آنها ممکن است به مشاور ژنتیک ارجاع داده شوند تا درک بهتری از الگوی وراثت و خطر انتقال بیماری به فرزندان آینده داشته باشند.
۵. تستهای تکمیلی
- الکترواکیولوگرافی (EOG): برای ارزیابی عملکرد لایههای بیرونی شبکیه استفاده میشود.
- آزمایشهای حساسیت به کنتراست: برای بررسی توانایی چشم در تشخیص تغییرات کنتراست.
پیگیری و مانیتورینگ
- پیگیری منظم: بیماران مبتلا به RP باید به صورت منظم توسط چشمپزشک معاینه شوند تا پیشرفت بیماری و اثرات درمانهای مختلف مورد ارزیابی قرار گیرد.
درمان و مدیریت
یک بیماری ژنتیکی و پیشرونده است که درمان قطعی ندارد، اما روشهای مختلفی برای مدیریت و کاهش سرعت پیشرفت بیماری وجود دارد. این روشها شامل درمانهای پزشکی، تغییرات در سبک زندگی و استفاده از تکنولوژیهای کمکی میشوند.
درمانها و مدیریت
۱. مکملها و تغذیه
- ویتامین A: مصرف ویتامین A به برخی بیماران کمک کرده است تا سرعت پیشرفت بیماری را کاهش دهند. دوز مصرف باید تحت نظر پزشک باشد زیرا مصرف بیش از حد ویتامین A میتواند سمی باشد.
- اسید دوکوزاهگزانوئیک (DHA): یک نوع اسید چرب امگا-۳ که ممکن است به حفظ سلامت شبکیه کمک کند.
۲. تجهیزات کمکی و توانبخشی بینایی
- عدسیهای بزرگنمایی و تلویزیونهای مدار بسته: برای بهبود دید نزدیک.
- وسایل کمکی نوری: مانند عینکهای مخصوص برای افزایش کنتراست و کاهش تابش نور.
- تجهیزات الکترونیکی: دستگاههای خوانشگر صفحه و نرمافزارهای بزرگنمایی.
۳. درمانهای ژنتیکی و سلولی
- تراپی ژنی: پژوهشهای اخیر روی تراپی ژنی متمرکز شدهاند که میتواند ژن معیوب را تصحیح یا جایگزین کند. برخی از این روشها هنوز در مراحل آزمایشی هستند.
- پیوند سلولهای بنیادی: پژوهشها نشان دادهاند که سلولهای بنیادی ممکن است بتوانند سلولهای شبکیه آسیبدیده را ترمیم کنند.
- ایمپلنتهای شبکیه: دستگاههایی که میتوانند جایگزین سلولهای آسیبدیده شبکیه شوند و سیگنالهای نوری را به سیگنالهای الکتریکی تبدیل کنند که توسط مغز تفسیر میشوند.
۴. درمانهای دارویی
- آنتیاکسیدانها: داروهایی که حاوی آنتیاکسیدانها هستند ممکن است به کاهش آسیب اکسیداتیو در شبکیه کمک کنند.
- داروهای محافظ شبکیه: برخی داروها ممکن است به حفاظت از سلولهای شبکیه در برابر آسیبهای بیشتر کمک کنند.
۵. مراقبتهای حمایتی
- مشاوره ژنتیک: برای فهم بهتر الگوی وراثت بیماری و مشاوره درباره خطرات انتقال به نسلهای بعدی.
- حمایت روانی: دریافت حمایت روانی و مشاوره میتواند به بیماران کمک کند تا با چالشهای ناشی از این بیماری بهتر کنار بیایند.
۶. پیشگیری از عوارض
- محافظت از چشمها در برابر نور شدید: استفاده از عینکهای آفتابی با فیلتر UV برای کاهش آسیب ناشی از نور خورشید.
- معاینات منظم: مراجعه منظم به چشمپزشک برای مانیتورینگ پیشرفت بیماری و تطبیق برنامه درمانی.
نتیجهگیری
در حال حاضر هیچ درمان قطعی برای رتینیت پیگمانتوزا وجود ندارد، اما با استفاده از روشهای مدیریتی مختلف و پیشرفتهای پژوهشی، میتوان کیفیت زندگی بیماران را بهبود بخشید و سرعت پیشرفت بیماری را کاهش داد. بیماران باید تحت نظر متخصصین چشم و ژنتیک باشند و از آخرین پژوهشها و درمانهای موجود بهرهمند شوند.
سلامت باشید.